


Искусственный интеллект решил задачу, которая уже около полувека остается одной из самых актуальных для биологии: предсказание третичной структуры белков по первичной. Теперь, зная аминокислотную последовательность крупной белковой молекулы, можно будет рассчитать ее трехмерную пространственную конфигурацию. О достижении сообщается в пресс-релизе, распространенном британским стартапом DeepMind.
Дело в том, что свойства и функции белков определяются их трехмерной структурой, и многие важные находки о том, как именно они работают, сделали на основе именно таких структур. Десятилетиями для этого применяют такие методы, как рентгеновская кристаллография, ядерный магнитный резонанс или криоэлектронная микроскопия — длительные, сложные и трудоемкие. Однако даже они не всегда справляются; в результате сегодня установлены 3D-структуры примерно 170 тысяч белков из около 200 миллионов, известных науке.
Между тем в природе третичная структура белков определяется первичной — последовательностью аминокислот, которые образуют цепочки этих молекул: они складываются естественным образом, сами по себе. Этот процесс называют «укладкой», фолдингом белка. Неудивительно, что долгие годы ученые стремились к тому, чтобы моделировать его математически. Задача оказалась настолько сложна, что даже применение суперкомпьютеров здесь не слишком помогло: число вариантов, которые требуется рассчитать для молекул, состоящих из сотен аминокислот, получается астрономическим.

Чтобы стимулировать работы в этом направлении, с 1994-го каждые два года проводят испытания CASP (Critical Assessment of protein Structure Prediction — «Критическая оценка предсказания структуры белков»). Создатели подобных проектов и алгоритмов со всего мира получают аминокислотные последовательности примерно сотни белков, структуры которых еще неизвестны, и пытаются вычислить их с помощью своих моделей. Параллельно в лабораториях работают ученые, действующие «классическими» методами структурной биологии. Затем полученные структуры сравнивают, вычисляя величину совпадения — GDT.
Значения GDT от 90 до 100 считают точным предсказанием структуры, и для коротких пептидов, состоящих из нескольких десятков аминокислот этого удалось добиться еще в 1990-х. Однако для белков, включающих сотни аминокислот, GDT долгие годы держались на «позорном» уровне — около 20. Лишь несколько лет назад, используя сложнейшие алгоритмы, эту цифру удалось довести до 40, чего по-прежнему недостаточно.

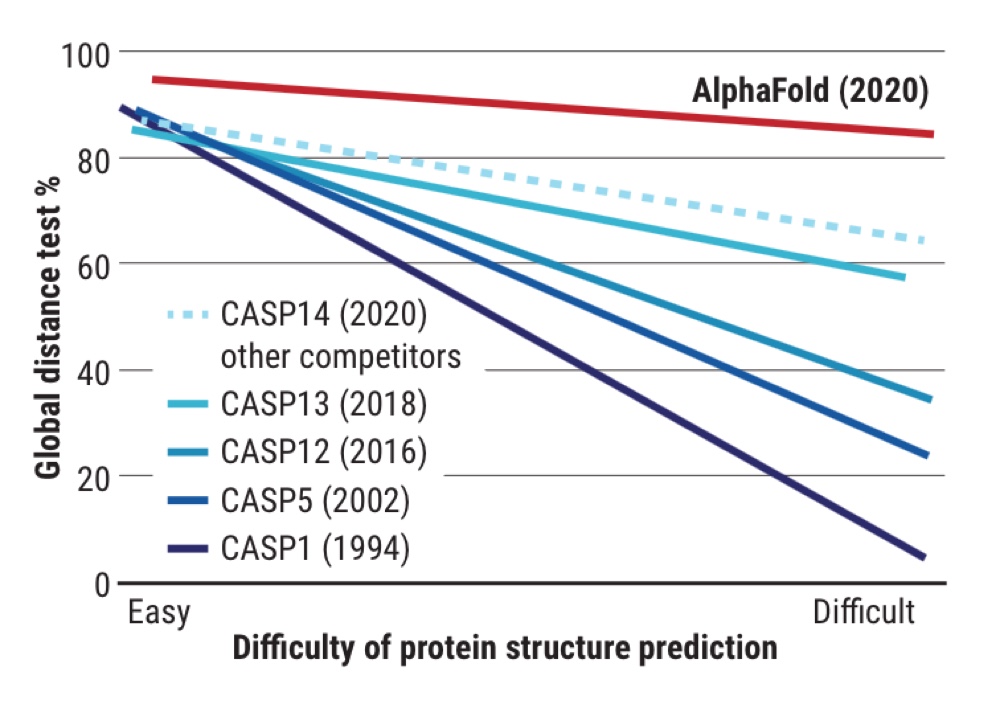
С 2018 года в конкурсе CASP участвует и проект AlphaFold, который разрабатывает британская компания DeepMind — та самая, в которой ранее создали ИИ, побеждающий профессионалов игры го. Уже тогда AlphaFold оказался лидером рейтинга, продемонстрировав GDT на уровне до 60 даже для сложнейших структур. К конкурсу 2020 года ИИ усовершенствовали и обучили на 170 тысячах известных белковых структур. На испытаниях он оказался способен предсказывать фолдинг со средним GDT более 92 и свыше 87 — для наиболее сложных молекул.
Специалисты уже называют это событие одним из важнейших прорывов последних лет. Возможно, вскоре нейросети позволят рассчитывать трехмерные структуры белков «на лету», по мере необходимости. Задача, которая прежде была настолько сложна, что авторы некоторых таких работ удостаивались Нобелевской премии, станет рутиной.