


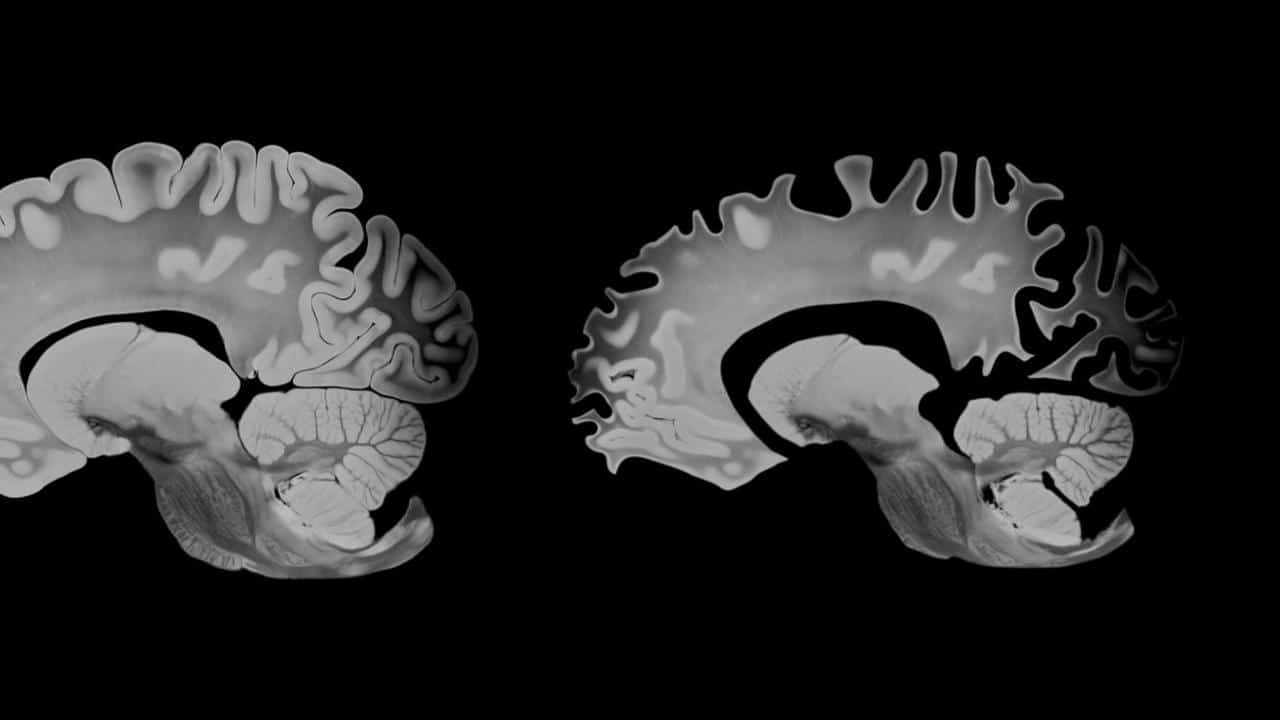
Болезнь Альцгеймера традиционно связывают с накоплением бляшек белка амилоида и клубков тау-белка в тканях головного мозга. В последние годы, однако, множество исследований показало, что именно скопления тау-белка лучше коррелируют с ухудшением памяти и когнитивных функций. Выяснилось, что в мозге пациентов тау-белок формирует парные спиральные филаменты (paired helical filaments, PHF), которые затем собираются в нейрофибриллярные клубки (neurofibrillary tangles, NFT). При этом ученым было неясно, что именно лежит в основе перехода нормального тау-белка в патологическую форму.
Теперь исследовательская группа из Колумбийского университета (США) обнаружила, что ведущую роль в запуске болезни Альцгеймера может играть особая система контроля качества белков, которая присутствует только в нейронах. Эту структуру ученые назвали «нейропротеасома» (neuroproteasome).
Поясним: обычная протеасома действует как клеточная система утилизации, расщепляя поврежденные или ненужные белки. Несколько лет назад у нейронов обнаружили ее дополнительный вариант. Располагаются нейропротеасомы непосредственно на внешней мембране нервных клеток и специализируются на уничтожении недавно синтезированных белков, которые особенно подвержены ошибкам сворачивания.
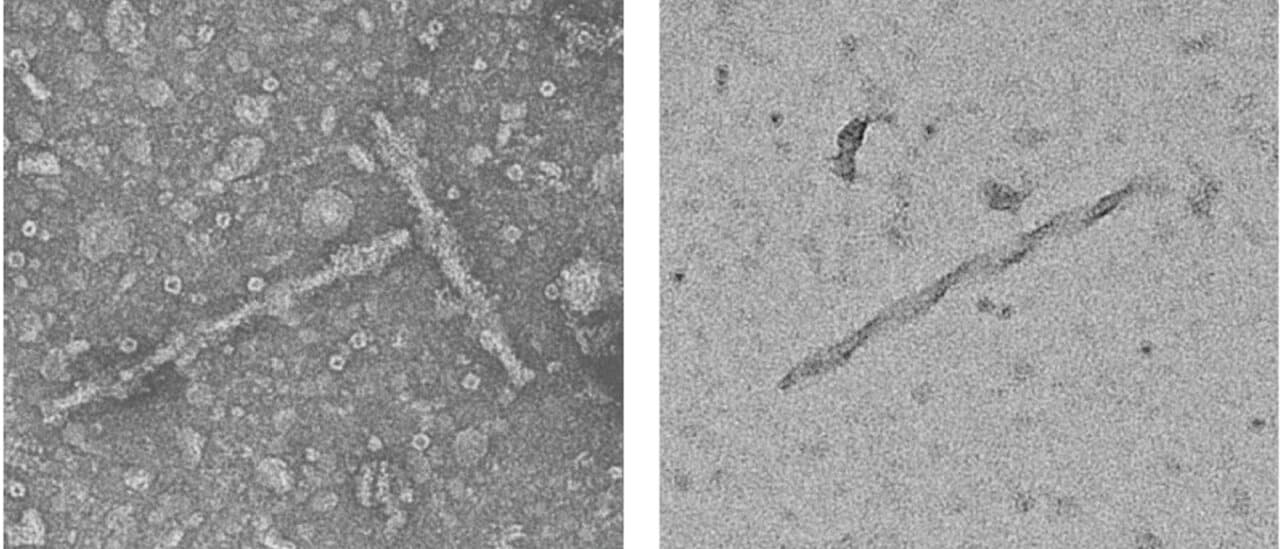
Чтобы выяснить функцию этой необычной структуры, нейробиологи разработали инструменты, позволяющие избирательно блокировать работу нейропротеасом. В результате уже вскоре после подавления их активности в нейронах начинали формироваться PHF. Причем по своим биохимическим свойствам и микроскопической структуре они были очень похожи на те, что находят в мозге пациентов с болезнью Альцгеймера.
Эксперименты проводились как на культурах нейронов, так и на мышах. Во всех случаях нарушение работы нейропротеасом приводило к самопроизвольному образованию патологических скоплений тау-белка. Это важно, поскольку большинство существующих моделей болезни Альцгеймера основаны на искусственном «заражении» животных человеческим PHF и другими патологическими формами тау-белка, полученными из тканей пациентов или выращенными в лаборатории. Это мешает изучать самые первые этапы развития недуга.
Особое внимание ученые обратили на ген APOE — крупнейший известный генетический фактор риска болезни. Ген существует в трех основных вариантах: APOE2, APOE3 и APOE4, которые кодируют разные формы белка ApoE. У носителей APOE4 риск возникновения патологии существенно выше, тогда как вариант APOE2 считается защитным.
Выяснилось, что разные формы белка ApoE по-разному влияют на количество нейропротеасом на поверхности нейронов. Наибольшее их число наблюдалось у носителей ApoE2, промежуточное — у ApoE3, а минимальное — у ApoE4. Именно по этой причине нейроны с вариантом гена APOE4 были более уязвимыми к нарушениям системы утилизации белков и быстрее начинали накапливать патологические формы тау-белка.
Эту закономерность подтвердили, дополнительно проанализировав ткани человеческого мозга. У людей с двумя копиями APOE4 количество нейропротеасом было много ниже, чем у носителей других вариантов гена. Кроме того, оказалось, что число нейропротеасом постепенно уменьшалось с возрастом. Поскольку старение остается главным фактором риска, результаты помогают объяснить, почему вероятность развития заболевания резко возрастает в пожилом возрасте.

Таким образом ученые связали в единую картину сразу два важных фактора недуга — старение и APOE4. Их модель показала, что возрастное снижение числа нейропротеасом и генетически обусловленный дефицит этих структур ослабляют контроль над качеством белков в нейронах. По итогу вероятность неправильного сворачивания тау-белка возрастает, запуская процесс формирования PHF, характерных для болезни Альцгеймера.
Результаты научной работы, опубликованной в журнале Nature Neuroscience, открывают перспективное направление для исследований. Если ученым удастся разработать способы поддержания или усиления работы нейропротеасом, это позволит предотвращать образование тау-клубков до развития необратимой гибели нейронов и появления симптомов деменции.