


Результаты опубликованы в журнале Physical Review B. В последние годы активно развивается разработка машинно-обучаемых межатомных потенциалов. Они способны обеспечить быстроту и точность моделирования структуры и свойств материалов. Квантово-механические методы, например, теория функционала плотности дают высокую точность вычислений, однако требуют значительных вычислительных ресурсов и времени. Машинное обучение ускоряет вычисления больших систем, практически не уступая в точности. Одна из острых проблем в применении машинного обучения заключается в обеспечении физической достоверности.
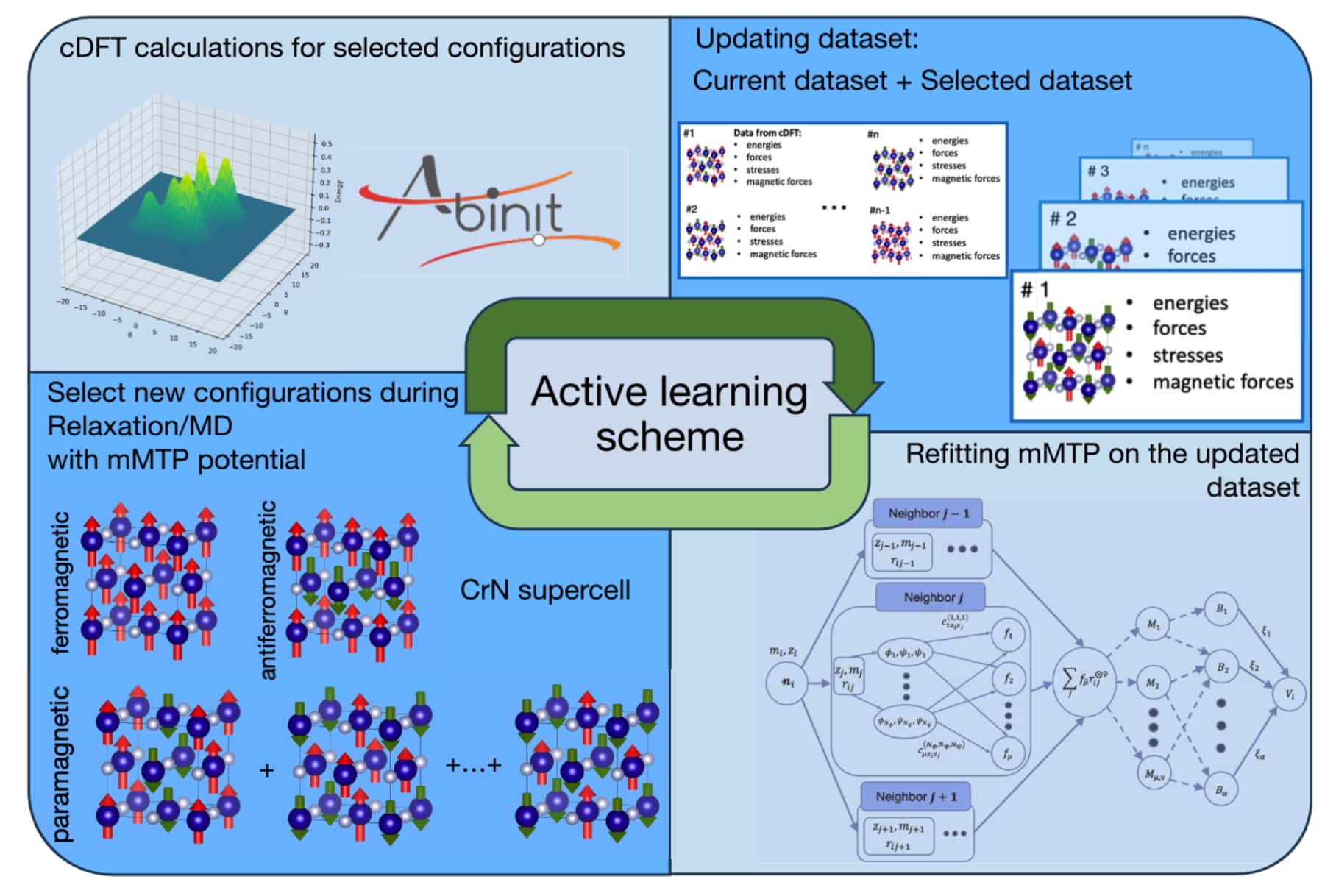
В своей новой работе ученые из МФТИ, Сколтеха, ВШЭ и их иностранные коллеги предложили алгоритм автоматического обучения машинно-обучаемого межатомного потенциала с магнитными степенями свободы. Он ускоряет трудоемкие квантово-механические расчеты при исследовании парамагнитных материалов, при этом сохраняя их высокую точность.
Магнитные моменты становятся новой переменной, что усложняет обучение потенциала. Процесс моделирования с использованием магнитного межатомного потенциала состоит из двух этапов. На первом этапе оптимизируется величина магнитных моментов при фиксированных координатах атомов и параметрах решетки так, чтобы полная энергия системы была минимальна. На втором этапе магнитные моменты фиксируются и выполняется молекулярно-динамическое моделирование, в ходе которого изменяются координаты атомов и параметры решетки с учетом магнитного взаимодействия.
Кроме того, наличие магнитных моментов в функциональной форме потенциалов усложняет его обучение. Для решения этой задачи исследователи разработали алгоритм, который автоматически выбирает оптимальные конфигурации для обучающей выборки. Алгоритм отслеживает конфигурации, возникающие прямо в процессе моделирования с обучаемым потенциалом, и для отобранных конфигураций проводятся расчеты с помощью теории функционала плотности. Полученные данные добавляются в обучающую выборку, на основе которой происходит обучение потенциала.
«Главной особенностью разработанного нами потенциала является возможность отбора конфигураций прямо во время моделирования с обучаемым потенциалом, например, в ходе молекулярной динамики. Таким образом, появляется возможность автоматизировать процесс составления обучающей выборки, так как потенциал сам отбирает релевантные конфигурации для последующего их расчета с помощью теории функционала плотности и дообучения на них. Еще одной особенностью является учет магнитных моментов конфигураций при отборе в ходе активного обучения», — рассказал Иван Новиков, доцент факультета компьютерных наук НИУ ВШЭ, доцент кафедры химической физики функциональных материалов МФТИ, старший научный сотрудник Сколтеха.
Ученые протестировали новый подход на материале CrN с кубической кристаллической решеткой, подобной кристаллической решетке поваренной соли. Свойства этого материала хорошо известны, и поэтому он позволил определить надежность разработанного подхода. Кроме того, особенностью данного материала является то, что при температурах выше комнатной он находится в парамагнитном состоянии, что являлось дополнительным усложнением апробации предложенной методологии. Результаты показали, что алгоритм точно воспроизводит константы упругости и термические свойства. Рассчитанные фононные спектры согласуются с экспериментальными данными. Ученые отмечают, что разработанный алгоритм универсальный и его можно применять для других материалов.
Итак, предложенный подход показал высокую точность в воспроизведении механических, динамических и термических свойств парамагнитного CrN, демонстрируя ресурс для широкого применения в материаловедении.
«Мы планируем добавить неколлинеарный магнетизм в функциональную форму нашего потенциала. Также мы хотим разработать и апробировать метод предсказания температуры перехода в парамагнитное состояние с использованием метода Монте–Карло с переворотом магнитных моментов в ходе молекулярной динамики», — поделился планами на дальнейшие исследования Иван Новиков.
В работе участвовали ученые из МФТИ, Сколтеха, НИУ ВШЭ, Института химии твердого тела и механохимии СО РАН, Института биохимической физики имени Н. М. Эмануэля РАН, Института Материаловедения Кальяри (Италия), Центра материалов Леобен (Австрия).